Enantioselective Total Synthesis of (-)-Lansai B and (+)-Nocardioazines A and B
Haoxuan Wang and Sarah E. Reisman¶*
Angew. Chem. Int. Ed. 2014, 53, 6206-6210 DOI: 10.1002/anie.201402571
Nothing incites apoplexy in your favorite peptide chemist like mentioning how “real” synthetic chemists are dismissive of the challenges that can arise, from time to time, in the formation of amide bonds. C’mon. How hard can it really be? Activate an acid using your favorite alphabet soup reagent and allow an amine nucleophile to take it from there. Done…. But sometimes things get complicated. Sarah Reisman’s#^ hot-off-the-presses Angewandte Chemie communication reporting the syntheses of (-)-lansai B (1) and the “no cardio required” (+)-nocardioazines A & B (2 & 3) reveal that even a card-carrying synthetic chemist can be stymied by amide bond formation, for a while. As it turns out, the forces of amide bond formation were used for good – in the macrocyclization of (+)-nocardioazine B.
This post is the second installment of an annual exercise for my organic synthesis class where I try to deconstruct a total synthesis communication paragraph by paragraph. The strategy and execution of the writing are put on equal footing with that of the chemistry. Our goal is to understand how the manuscript – both its text and its graphics – communicates the ideas behind the science and the agenda/priorities of the author. It is a case study in synthesis AND manuscript writing. The discussion of the manuscript in class emphasizes active reading and what behaviors active reading entails.
So there’s no better place to start than at the beginning. The title “Enantioselective Total Synthesis of (-)-Lansai B and (+)-Nocardioazines A and B” and abstract are straightforward stylistically and they give clear clues about the story that will unfold in the manuscript. Three words/phrases do the work that is needed here. 1 – The term “enantioselective” indicates how the chirality of the natural products will be introduced in the molecule: via chiral catalysts in enantioselective reactions in the synthetic sequence. 2 – “Formal [3+2] cycloadditions” refers to a new and powerful reaction introduced earlier by the group for formation of pyrroloindolines. 3 – By noting that macrocyclization occurred by “intramolecular diketopiperazine formation” the authors tip their hand on the ultimate route for the synthesis of nocardioazine A. Now to the manuscript proper.
Paragraph 1: The introduction is textbook for a synthesis paper. It gives a quick one-two punch in the form of biological activity (1, 2, and 3 in Figure 1 are potential antibacterial and anticancer agents.) and a description of the structure (They are bis(pyrroloindolines) linked via a diketopiperazine.). It then back-fills with some details for each point. First, the star compound, (+)-nocardioazine A (2), is a P-glycoprotein inhibitor. P-glycoprotein is an efflux pump central to multi-drug resistance resistance in several cancers. The remainder of the paragraph points out the subtleties of the structures, especially the stereochemical relationship between the group at the C3 of the pyrroloindoline unit and the carboxamide, giving rise to the exo- and endo- nomenclature throughout the paper. (-)-Lansai B (1) is constructured of two exo pyrroloindolines in the same enantiomeric series, whereas nocardioazines A (2) and B (3) are made from one endo- and one exo- pyrroloindoline of opposite enantiomeric series. Compounds 4–7 in Figure 1 nicely illustrate the relationships. Reisman emphasizes that these precursors make “appealing synthetic targets for asymmetric catalysis where selection of the appropriate enantiomer of the catalyst dictates the absolute stereochemistry of the pyrroloindoline building block.” That’s a great sentence. She’s building up the value of their approach before she fully reveals it. The paragraph closes by noting that there is one reported synthesis of 3 and none of 1 and 2.
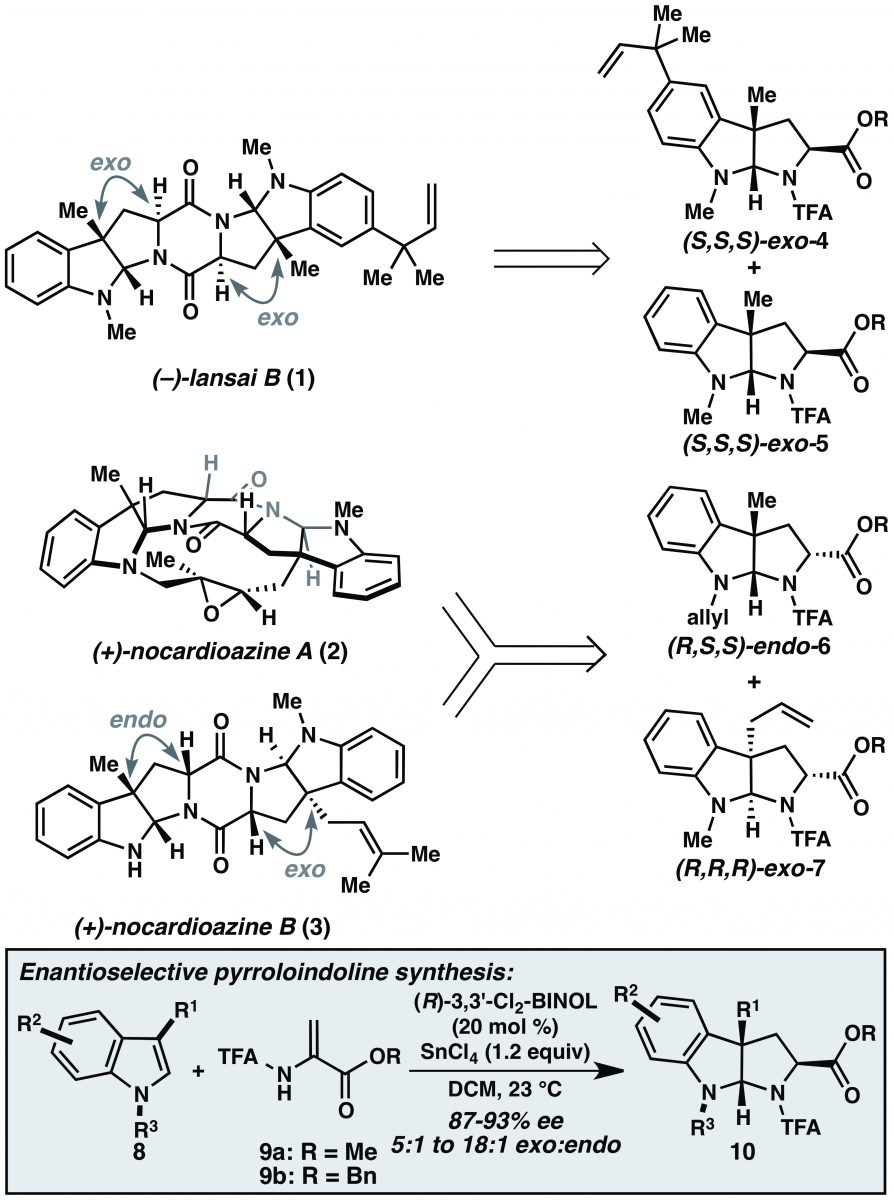
Paragraph 2: Some readers may have realized already that this synthesis will showcase the formal (3+2) cycloaddition methodology based on the hints (really statements) in the abstract and first paragraph. The pyrroloindoline natural product targets in the this paper, among others, likely motivated the development of the transformation. That’s a standard argument made by organic chemists for the development of new reactions. If you didn’t see it coming, it becomes explicit in the first sentence of this paragraph. The method for enantioselective synthesis of pyrroloindolines (box in Figure 1) is a major component of their retrosynthetic analysis. In fact, that IS their retrosynthetic analysis as shown in the figure. Reisman trusts that you can make the DKP disconnection and, if you don’t see it now, she’ll walk you through it shortly. “Utility” in the last sentence of the paragraph rings in your ears – this report on total syntheses exclaims the utility of the formal (3+2) methodology.
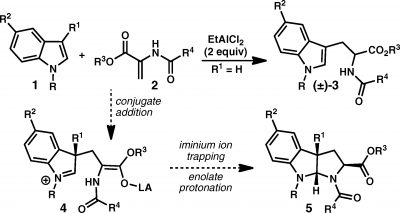
Despite the centrality of the reaction to the syntheses, it gets a mere mention and a call of Figure 1 but that’s nearly it as far as explanation. The reader must work through the figure and, if necessary, revisit reference 4a, which is the JACS communication that describes the reaction. The reaction uses C3 substituted indoles and 2-amidoacryates in the presence of SnCl4 and catalytic (R or S) BINOL to deliver pyrroloindolines with high enantioselectivity. Note the cis relationship between the R1, H, and carboxylate groups in structure 10 of Figure 1; this is the exo- relationship defined earlier. The reaction is formally equivalent to a (3+2) cycloaddition where the two olefinic carbons and the nitrogen of the amidoacrylate are the “3” and the two carbons of the indole are the “2”. Scheme 1 from the JACS communication gives some insight into the transformation and to the origin of the enantioselectivity. Conjugate addition by the indole onto the amidoacrylate is followed by protonation of the resulting enolate followed by ring closure. With regard to selectivity, Reisman speculates in the JACS communication, “[conjugate addition] occurs with modest levels of catalyst control, whereas the enolate protonation step occurs with high levels of catalyst control. In effect, the catalyst-controlled protonation step serves to resolve the mixture of enantiomeric intermediates.”
With that, they’re finished with introductions and on to the business of the syntheses.
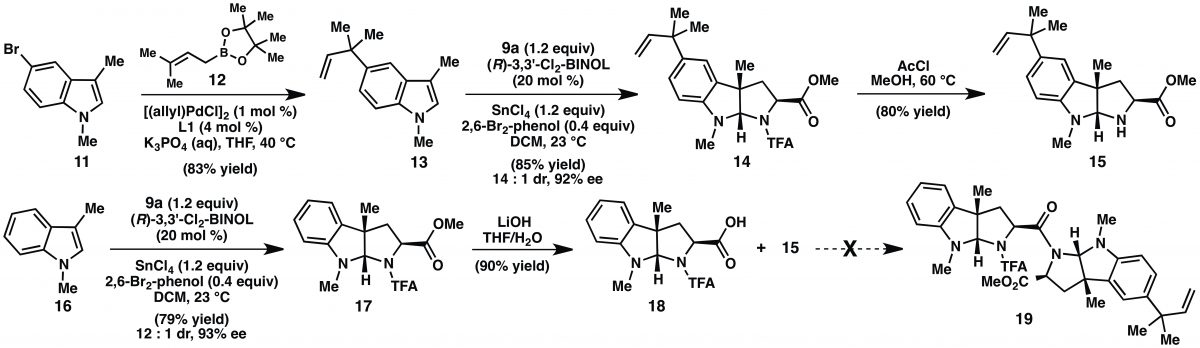
Paragraph 3: Like a lion after a young gazelle, Reisman aims for (-)-lansai B (1) as her first target. Here she provides a little more detail on the retrosynthesis by stating that formation of the DKP would be the way to unite the two pyrroloindoline fragments, 4 and 5, via “sequential amide bond formation.” Simple. She finishes the paragraph by calling the starting materials for the synthesis of 4 and 5, indoles 13 and 16, both of which are in Scheme 1. If you wondered where the indoles themselves came from, reference 4a is there to guide you. The SI of that paper shows a tandem amination-Heck reaction with substituted bromo-iodo-benzenes and allyl amine to deliver the indoles.
From here, the organization of the paragraphs communicating the syntheses of 1–3 largely follows the synthetic strategy. That is, Reisman walks the reader through pyrroloindoline synthesis using the formal (3+2) method and then make use of those advanced intermediates in the formation of DKPs to complete the syntheses. As always, there are minor twists and turns in the storyline though.
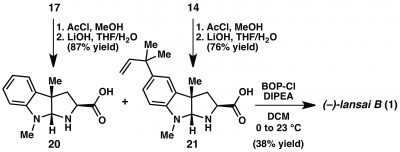
Paragraph 4: This paragraph takes the reader through the synthesis of two pyrroloindolines, 15 and 18, from the corresponding indoles. Again calling Scheme 1, the paragraph starts by describing a Suzuki-Miyaura reaction that installed the prenyl group on intermediate 15. To be clear, this group is the only difference in the two pyrroloindoline units in (-)-lansai B (1). “Subjection” of 13 to the cycloaddition reaction provided 14 in 85% yield, 14:1 dr, and 92%ee. An aside: Subjection is a great word – it’s perfectly descriptive but it bears a tinge of that, “Is it really a word?” feel to it. Anyway, on scale up (> 1.0 mmol), she notes that lower yields were obtained and that a scan of protic additives showed that addition of 2,6-dibromophenol maintained yield and selectivity. They suggest that, “…2,6-dibromophenol facilitates turnover of the chiral catalyst, but is not reactive enough to protonate the transient enolate directly in a nonselective fashion.” It’s like a goldilocks acid in a milieu of acids in the cycloaddition reaction. This observation underscores the complexity of the reaction mechanism. Thankfully, the results are easily understood and useful. Conversion of 16 to 17 follows the same protocol (79%, 12:1 dr, 93% ee). The paragraph continues on with some deprotections to end with intermediates 15 and 18. (-)-Lansai B (1) is about to yield DKP formation.
Paragraph 5: The concession paragraph. The plan was to finish (-)-lansai B (1) through DKP formation by sequential amide bond formation. Preparation of intermediate 19 in Scheme 1 was to be the first of the two amide bond forming reactions. Despite scanning a “wide variety of peptide coupling conditions” – queue visions of alphabet soup – they did not see coupling but only decomposition of 18. Reisman then recalls a successful coupling of exo-pyrroloindolines by Danishefsky and coworkers in their synthesis of amauromine. There both nitrogens were protected as t-butyl carbamates whereas 18 has one N-alkyl and one N-trifluoracetamide. She comments, “these findings reveal that the N substitution of the exo- pyrroloindoline significantly influences the stability of the activated ester under peptide coupling conditions.” So peptide chemistry can pose challenges. They then present an alternative strategy involving the completely deprotected amino acids 20 and 21 (Scheme 2). The plan is to take a hit on yield and mix 20 and 21 to form three DKPs in one reaction; in their estimation this was more efficient that several protecting group manipulations. Coupling using BOPCl gave the two homodimers, each in 20% yield, and (-)-lansai B (1) in 38%; the synthesis was six steps with an overall yield of 20%. In hindsight, the concession was a small one and they learned a little about the amide couplings along the way.
Paragraph 6: On to the nocardios! Paragraph 6 revisits Figure 1 as it presents the retrosynthesis of 2 and 3 in greater detail. Again the strategy makes use of DKP formation but now targets the more complicated pyrroloindolines 6 and 7 as key intermediates. Compound 6 is the sole endo– configured pyrroloindoline in the paper. Note that its C2 and C3 positions are of the same enantiomeric series as 4 and 5, though. Access to it will require cycloaddition as before followed by epimerization of the carbon a- to the carboxylate. Exo-pyrroloindoline 7, on the other hand, is of the opposite enantiomeric series relative to 4 and 5 and it contains an allyl group (rather than methyl) at C3. Its synthesis will be a real test of the formal (3+2) cycloaddition methodology. The paragraph continues down the forward synthetic path by presenting the cycloaddtion reaction of 22 (Scheme 3) with 2-amidoacrylate 9a to give 23. For the preparation of 23, S rather than R-BINOL is used; this is exactly the point of using asymmetric catalysis and nicely illustrates the power of the cycloaddition. They take a bit of hit on yield for this transformation though. They obtain 23 in 52% yield, 19:1 dr, and 90% ee. The modest yield is attributed to an unwanted side product that results from allyl migration (from C3 to C2 of the indole) under the reaction conditions. However, the presence of the allyl group challenges the scope of the cycloaddition beyond what was done previously and fares well overall. Deprotection of the TFA group then gives one coupling partner, exo-24.
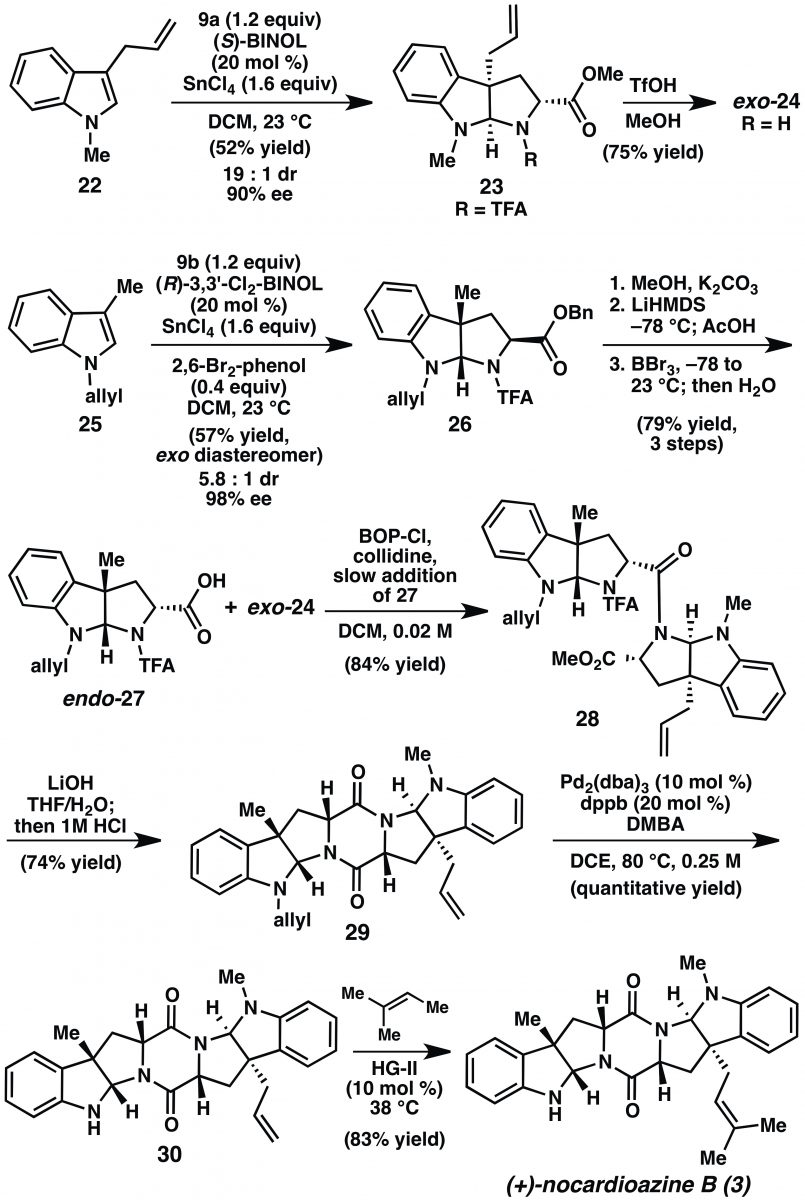
Paragraph 7: Paragraph 7 contains more pyrroloindoline synthesis. This time it’s N-allyl-C3-methyl-indole 25 that is the starting material; this indole is also a little more complicated than the models used in the development of the method. Of note is that 9b, the benzyl ester version of the 2-amidoacrylate, is used in the cycloaddition reaction (Scheme 3). This is the only instance among the four cycloadditions that uses this acrylate. It’s not discussed, but in the methodology paper, this acrylate gave the best combination of yield, dr and ee. It is up to the reader to surmise that despite the modest yield (due to lower diastereselectivity in this case), 9b must have given better results than 9a for the transformation. Transesterification, the need for which is not really explained, sets up the epimerization of the carbon alpha to the carboxylate of 26. Cleavage of the methyl ester then reveals acid endo-27 which is ready for amide bond formation.
Paragraph 8: The nocardio A/B synthesis is now at the stage where pyrroloindolines 24 and 27 are “poised” for DKP formation. Coupling in this case using BOPCl was successful. The product, 28 (Scheme 3), was isolated in 84% yield. A footnote directs the reader to the SI for details of the optimization of the yield of the reaction at this point. Inspection of the SI shows that the base, equivalents of amine (24), and rate of addition of acid (endo-27) to the mixture were optimized. She then pays additional homage to the amide bond by comparing the failure to couple 15 and 18 with the success with 24 and 27, writing, “…in addition to the N-substituents, the relative stereochemistry of the pyrroloindoline coupling partners is determinate of the ease of peptide formation.” A saponification-acidification sequence closes the DKP to give 29 (74%). Removal of the N-allyl group then provides 30, a versatile intermediate common to both nocardio A and B. The paragraph ends with the efficient cross methathesis of 30 and 2-methyl-2-butene. The reaction completes the (+)-nocardioazine B (3) synthesis; their route delivered it in 21% overall yield in 9 steps. Modestly, the paper defers the opportunity to compare this synthesis of 3 to the one reported by the group of Ye (10 steps, 12%).
Paragraph 9: Reisman’s attention now turns to (+)-nocardioazine A (2). The plan is to advance compound 30, the penultimate intermediate in the synthesis of (+)-nocardioazine B (3). Cross metathesis of 30 with methacrolein, Luche reduction, and mesylation-chlorination proceed without incident to give 33 (Scheme 4). “Gratifyingly,” (Another great word, although I would have stolen a Wood/Stoltz gem, “To our delight,”) macrocyclization via N-alkylation was also successful, giving 34 in 73% yield. This intermediate is so close to the target itself; it is amongst those “lofty heights of an advanced intermediate”. All that’s left is a seemingly mundane epoxidation that should be quite diastereoselective based on the shape of the macrocycle. The writing seems down right despondent when it lists the epoxidation conditions that yielded little more than the unstable N-oxide 35. Upon inspection of the solid state structure of 34, the recalcitrance of the trisubstituted double bond toward epoxidation was argued not to be steric, but rather resulted from the “electron-withdrawing nature of the allylic nitrogen.” No citation, no further explanation. The lack of additional discussion here leaves the reader grasping for a complete understanding of the lack of reactivity.
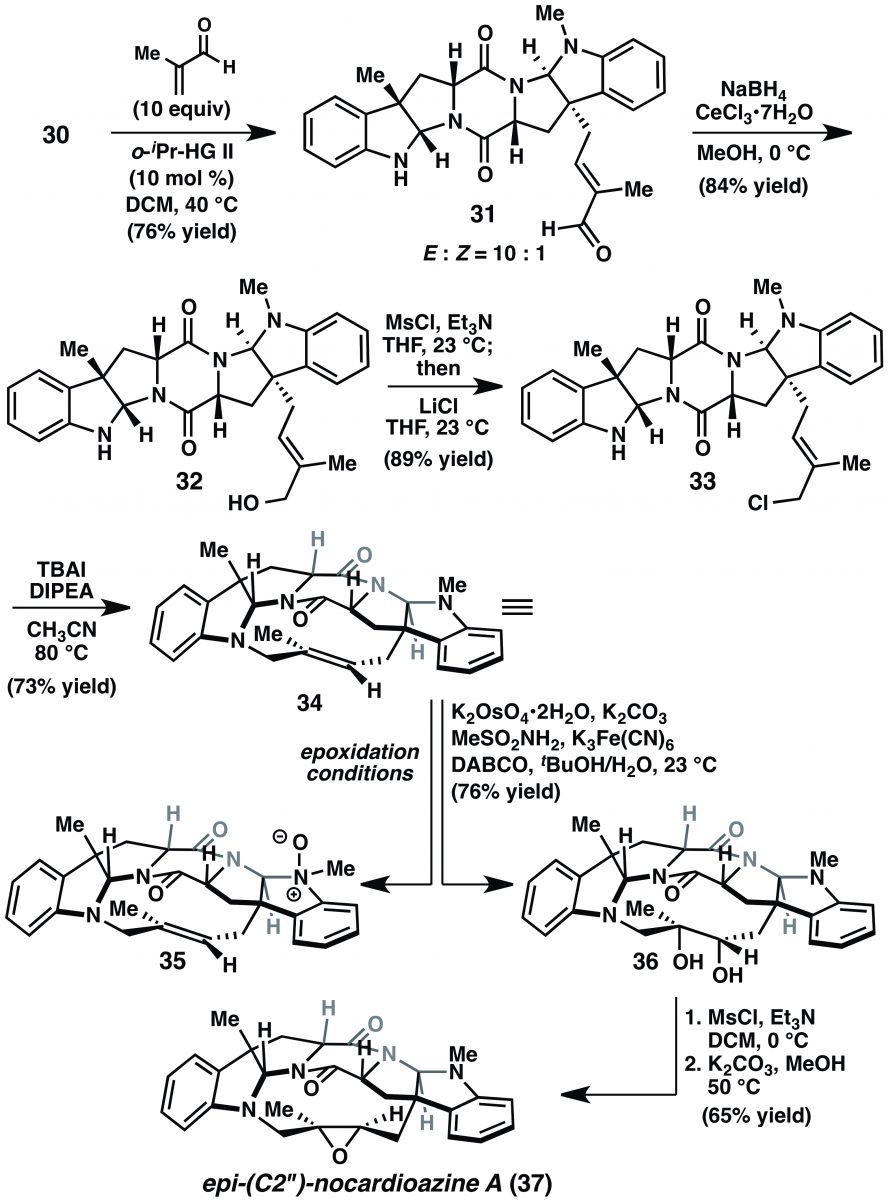
Paragraph 10: This paragraph details one last gasp to push intermediate 34 over the line to (+)-nocardioazine A (2). Dihydroxylation and chemoselective mesylation followed by intramolecular epoxidation delivered the C2’’-epi-nocardioazine A (37). Ultimately this was a dead end and Reisman moves on without any additional comment. Damn.
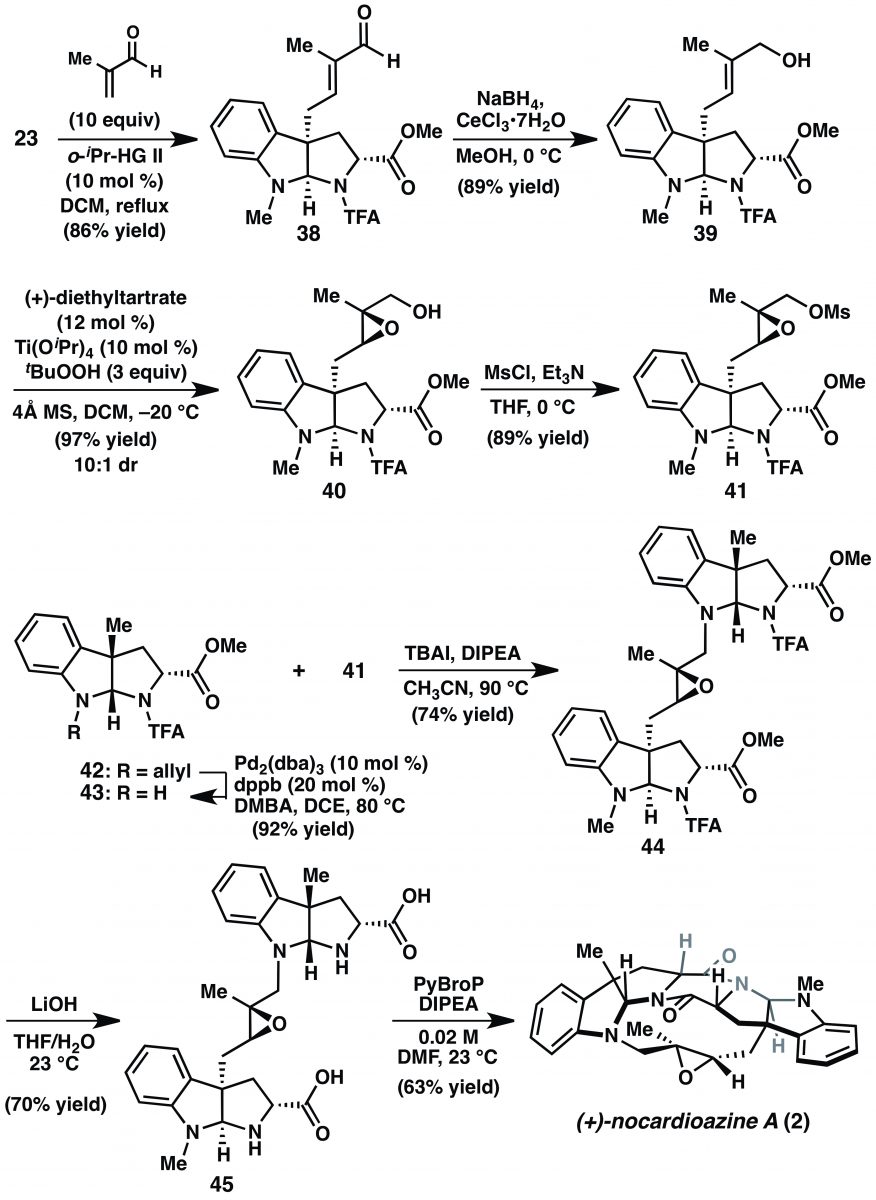
Paragraph 11: But then she fires it up one more time. Paragraph 11 walks through a revised synthesis of (+)-nocardioazine A (2) in Scheme 5. They intercept an early intermediate from the previous (attempted) route, pyrroloindoline 23, and proceed with a cross metathesis using methacrolein, Luche reduction and then Sharpless epoxidation on the resultant allylic alcohol giving intermediate 40. This was converted to the corresponding mesylate (41) in anticipation of the upcoming alkylation. Compound 43 is the amine nucleophile for the alkylation. It was prepared from 42 via de-allylation. Although not numbered there, 42 was also an intermediate in Scheme 4. The conversion of 26 to endo-27 entailed transesterification, epimerization, and demethylation to reveal the acid. Drop the last (demethylation) step and you’ve got 42. Alkylation of 41 with 43 then proceeds efficiently and without incident. Saponification of the methyl esters and hydrolysis of the trifluoroacetamides of 44, the product of the alkylation, gave 45. Lofty heights. “We were pleased to find that subjection of 45 to PyBroP in DMF promoted intramolecular DKP formation to afford (+)-nocardioazine A (2).” [YAWP! Mic drop.] Rather than exclaim, Reisman chooses to be understated, cool. She moves on to reassign the stereochemistry of 2 relative to the isolation paper; the reassigned structure of 2 is consistent with Ye and coworker’s reassignment of 3. The paragraph ends with the usual metrics (nine linear steps, 11% overall yield) and the observation that the macrocyclization by DKP formation was effective.
Paragraph 12: The wrap up hits the points that have been collected along the manuscript. 1 – We made the three related bis(pyrroloindoline) DKP natural products. 2 – The enantioselective formal (3+2) cycloaddition is useful to total synthesis. And finally, validation their respect for the amide bond. 3- “In addition, subtle changes in the relative stereochemistry and nitrogen substitution patterns of pyrroloindolines were shown to significantly influence the ability to prepare bis(pyrroloindolines) by DKP formation.” This finish signals Reisman’s appreciation of the amide bond as a worthy adversary.
——-
¶Thanks to Sarah E. Reisman for sharing ChemDraw versions of the figures and schemes from the Angewandte paper.
# Professor Reisman has a tenuous connection to UConn Chemistry. Timo Ovaska, her undergrad research advisor, earned his PhD with Bill Bailey in our department. She then traveled west to New Haven to work in John Wood’s lab when he was still at Yale. Though Sarah isn’t a native nutmegger, we’d gladly claim her as our own; the (+)-nocardioazine B synthesis shows a good bit of Yankee ingenuity.
^ Although this post has been written as though Professor Reisman is the sole researcher and writer, it has been done for clarity only. It’s safe to say that she, like all faculty, values the scientific collaboration and training with post-docs, grad students, and undergrads as the most enjoyable part of the job.